





Als ST-Hebung (auch: ST-Strecken-Hebung) bezeichnet man eine Verlagerung des J-Punkts und der anschließenden ST-Strecke über die isoelektrische Linie im Elektrokardiogramm (EKG). Sie markiert den Übergang von der ventrikulären Depolarisation (QRS-Komplex) zur Repolarisation (T-Welle).[1] Dieser Befund weist typischerweise auf eine myokardiale Schädigung, Ischämie oder andere kardiale Pathologien hin. Als signifikant gilt eine ST-Hebung von ≥0,1 mV in mindestens zwei benachbarten Ableitungen (außer V2–V3) bzw. in den Ableitungen V2–V3 von ≥0,2 mV bei Männern unter 40 Jahren, ≥0,15 mV bei Männern ab 40 Jahren und ≥0,15 mV bei Frauen.[2]
In der klinischen Praxis ist die ST-Hebung vor allem mit dem ST-Hebungs-Myokardinfarkt (STEMI) assoziiert, einer schweren Form des akuten Koronarsyndroms infolge eines vollständigen oder nahezu vollständigen Verschlusses einer Koronararterie. Dies führt zu einer transmuralen Myokardischämie und bei ausbleibender rascher Therapie potenziell zu Myokardnekrose.[3] Die Diagnose eines STEMI setzt eine ST-Hebung im klinischen Kontext, beispielsweise bei thorakalen Schmerzen, meist innerhalb von 12 Stunden nach Symptombeginn voraus und wird häufig durch serielle EKGs sowie kardiale Biomarker wie Troponin untermauert.[2] ST-Hebungen können jedoch auch nichtischämische Ursachen haben, darunter Perikarditis (typischerweise diffuse, konkav ansteigende Hebungen), Frührepolarisationsmuster (benigne Variante mit J-Notching und aufsteigender ST-Strecke), linksventrikuläre Hypertrophie, Linksschenkelblock, Ventrikelaneurysma oder Brugada-Syndrom.[1][4]
Die klinische Relevanz der ST-Hebung liegt in ihrer Funktion als zeitkritischer Marker, der eine umgehende diagnostische Einordnung erfordert, um Notfallbilder wie den STEMI von Nachahmern abzugrenzen. Fehldiagnosen können zu unnötigen Interventionen oder gefährlichen Therapieverschiebungen führen.[5] Beim STEMI betonen Leitlinien die rasche Reperfusionsbehandlung, um den Myokardschaden zu begrenzen und die Überlebenswahrscheinlichkeit zu verbessern.[2] Insgesamt bleibt die korrekte Interpretation von Muster und Kontext der ST-Hebung im EKG essenziell für optimale Behandlungsergebnisse bei kardiovaskulären Notfällen.[5]
EKG-Grundlagen
Definition der ST-Strecke
Die ST-Strecke ist der Abschnitt des Elektrokardiogramms (EKG) zwischen dem J-Punkt, der das Ende des QRS-Komplexes markiert, und dem Beginn der T-Welle.[6][1] Sie entspricht dem Intervall zwischen dem Abschluss der ventrikulären Depolarisation und dem Beginn der relevanten ventrikulären Repolarisation.[1]
Die Quantifizierung der ST-Strecke erfolgt üblicherweise durch Messung ihrer Abweichung von der Grundlinie, die durch das PR-Segment definiert wird, in Millimetern.[7] Gemessen wird meist direkt am J-Punkt oder 60 Millisekunden danach (J+60 ms), insbesondere in Ableitungen mit geneigter ST-Strecke.[8][1]
Die Bezeichnung der ST-Strecke entstand im Rahmen des allgemeinen Benennungssystems der EKG-Wellen, das Willem Einthoven zu Beginn des 20. Jahrhunderts entwickelte, als er die Buchstaben P, Q, R, S und T auf aufeinanderfolgende Deflektionen in den galvanometrischen Aufzeichnungen anwandte.[9]
Im normalen EKG erscheint die ST-Strecke als flache oder leicht gekrümmte isoelektrische Linie auf Höhe der Grundlinie oder in unmittelbarer Nähe dazu.[6] Dieses Erscheinungsbild variiert je nach Ableitung: In den Extremitätenableitungen (I, II, III, aVR, aVL, aVF) ist sie typischerweise isoelektrisch, während sie in den Brustwandableitungen (V1–V6) eine geringe Konkavität oder minimale ansteigende Tendenz ohne pathologische Abweichung zeigen kann.[10][11]
Normale Muster der ST-Hebung
Normale ST-Hebungsmuster stellen benigne Varianten im EKG gesunder Personen dar. Am häufigsten handelt es sich um das Frührepolarisationsmuster (early repolarization pattern, ERP), das durch eine J-Punkt-Hebung mit nachfolgender konkav ansteigender ST-Strecken-Hebung gekennzeichnet ist, typischerweise unter 2 mm in den präkordialen Ableitungen V2–V4.[12][13] Häufig finden sich ein gekerbter oder verwaschener J-Punkt sowie prominente T-Wellen, was die Abgrenzung von pathologischen Veränderungen durch die stabile, aufsteigende Morphologie erleichtert.[12][14]
Das ERP findet sich besonders häufig bei jungen Männern, Leistungssportlern und Personen afrikanischer Abstammung. Studien zeigen für schwarze Männer eine Odds Ratio von 3,3 gegenüber anderen Gruppen; zudem ist das Muster bei Jugendlichen häufiger und nimmt mit steigendem Alter ab, sodass es nach dem 50. Lebensjahr selten wird.[15][16] Bei Sportlern reflektiert es einen erhöhten Vagotonus und Trainingseffekte und findet sich bei bis zu 20 % junger Erwachsener, ohne in asymptomatischen Fällen mit einem erhöhten Arrhythmierisiko einherzugehen.[16][17] Neuere Metaanalysen berichten Prävalenzen von bis zu 20–30 % bei Athleten und bis zu 48 % bei schwarzen Bevölkerungsgruppen.[18]
Eine Untergruppe des ERP kann eine gering ausgeprägte, diffuse ST-Hebung in mehreren Ableitungen ohne reziproke ST-Senkungen verursachen und dadurch die bei Perikarditis beobachteten Veränderungen imitieren, jedoch ohne begleitende klinische Zeichen wie ein Perikardreiben.[12][14]
Die Abgrenzung zur ischämischen ST-Hebung beruht auf dem Fehlen reziproker Veränderungen, einer konvexen Morphologie oder einer dynamischen Evolution. Beim ERP nimmt die Hebung unter Frequenzanstieg, etwa bei Belastung, typischerweise ab oder verschwindet, während ischämische Muster persistieren oder progredient sind.[19] Serielle EKGs mit zeitlicher Stabilität sprechen zusätzlich für eine benigne Genese und helfen, unnötige Interventionen zu vermeiden.[12]
Pathophysiologie
Elektrophysiologische Mechanismen
Die ST-Strecke im Elektrokardiogramm (EKG) entspricht der Plateauphase (Phase 2) des kardialen Aktionspotenzials, in der das Membranpotenzial durch ein Gleichgewicht zwischen einwärts gerichteten Calciumströmen (Ca²⁺; I_Ca) und auswärts gerichteten Kaliumströmen (K⁺), einschließlich des verzögerten Gleichrichterstroms I_K und des ATP-sensitiven K⁺-Stroms (I_K-ATP), nahe 0 mV gehalten wird.[20] Im normalen Myokard gewährleistet diese Plateauphase eine homogene Repolarisation über die ventrikulären Wandschichten, sodass die ST-Strecke isoelektrisch erscheint, weil während des Abschlusses der Repolarisation keine wesentlichen Spannungsgradienten bestehen.[21] Eine Ischämie stört dieses Gleichgewicht vor allem durch Verminderung von I_Ca infolge von ATP-Depletion und Azidose sowie durch Aktivierung von I_K-ATP, was insbesondere in epikardialen Zellen zu einer Verkürzung der Aktionspotenzialdauer (APD) führt.[22] Diese selektive APD-Verkürzung im Epikard erzeugt eine heterogene Repolarisation und führt in den darüberliegenden Ableitungen zur ST-Hebung.[20]
Transmurale Spannungsgradienten entstehen während der Ischämie, wenn ischämische epikardiale Regionen im ST-Intervall gegenüber gesünderen endokardialen Bereichen relativ depolarisiert bleiben und so Verletzungsströme erzeugen, die vom normalen (negativen) zum ischämischen (positiven) Gewebe fließen.[23] Diese systolischen Verletzungsströme, die experimentell erstmals quantifiziert wurden, manifestieren sich als ST-Hebung in Ableitungen über der ischämischen Zone, weil dort das extrazelluläre Potenzial positiv wird.[24] Bei schwerer transmuraler Ischämie verstärken Leitungsverzögerungen zwischen den Myokardschichten – etwa bis zu 87 ms im mittleren Myokard – diese Gradienten, verstärken den Stromfluss und erzeugen charakteristische „Tombstone“-ST-Hebungen.[20] Diastolische Komponenten tragen möglicherweise nur geringfügig bei; der zentrale Mechanismus ist die systolische Diskrepanz des Repolarisationszeitpunkts zwischen ischämischem und nichtischämischem Gewebe.[21]
Die Vektoranalyse von ST-Abweichungen interpretiert diese Verletzungsströme als resultierenden ST-Vektor, der zur epikardialen Oberfläche der ischämischen Region gerichtet ist; seine Größe spiegelt das Ausmaß der transmuralen Beteiligung wider.[25] Die ableitungsspezifische Projektion dieses Vektors erklärt, warum ST-Hebungen in benachbarten Ableitungen über dem Läsionsgebiet auftreten (positive Auslenkung), während in gegenüberliegenden Ableitungen reziproke ST-Senkungen zu beobachten sind (negative Auslenkung), und liefert damit eine räumliche Zuordnung der Ischämie.[25] So verschiebt eine anteriore Ischämie den ST-Vektor nach kranial und links, was zu ST-Hebungen in V2–V4 und zu ST-Senkungen in den inferioren Ableitungen führt.[20]
Tierexperimentelle Modelle haben diese Mechanismen durch kontrollierte Koronarokklusion aufgeklärt. In isolierten, arteriell perfundierten canine ventricular wedge preparations führt eine akute Ischämie zu einer Verkürzung der epikardialen APD und zum Verlust des Aktionspotenzialdoms, was direkt mit der ST-Hebung im transmuralen EKG korreliert.[22] Schweineherzmodelle mit regionaler Okklusion zeigen, dass Verletzungsströme innerhalb weniger Minuten entstehen, Repolarisationsgradienten verändern und zeitabhängige ST-Verschiebungen erzeugen, die extrazellulär gemessen werden können.[24] Weitere canine Okklusionsmodelle zeigen, dass die postokklusive Verlangsamung der transmurale Erregungsausbreitung die Spannungsgradienten verstärkt und damit die zentrale Rolle der heterogenen Repolarisation bei der Genese der ST-Hebung bestätigt.[20]
Ionische und zelluläre Grundlagen
ST-Hebungen entstehen durch Störungen der ionischen Ströme, die das kardiale Aktionspotenzial bestimmen, insbesondere auf zellulärer Ebene bei Ischämie. Im ischämischen Myokard führen eine extrazelluläre Akkumulation von Kalium (K⁺) und Wasserstoffionen (H⁺) zu einer partiellen Membrandepolarisation, wodurch spannungsabhängige Natriumkanäle (Na⁺) inaktiviert werden. Dies vermindert den maximalen Na⁺-Einstrom (I_Na), verlangsamt die maximale Anstiegsgeschwindigkeit des Aktionspotenzials (dV/dt_max) und beeinträchtigt die Erregungsleitung, besonders in epikardialen Regionen, wo dieser Effekt aufgrund einer negativeren Halb-Inaktivierungsspannung stärker ausgeprägt ist als im Endokard. Der daraus resultierende transmurale Spannungsgradient während der Systole trägt zur ST-Hebung bei, indem er im ischämischen Epikard relativ zum nichtischämischen Endokard eine Stromsenke erzeugt.[20]
Störungen der Calciumhomöostase verstärken diese ionischen Ungleichgewichte zusätzlich. Die ischämiebedingte intrazelluläre Azidifizierung aktiviert den Na⁺/H⁺-Austauscher, erhöht den Na⁺-Einstrom und führt über den reversen Na⁺/Ca²⁺-Austauscher zu einer intrazellulären Ca²⁺-Überladung. Diese begünstigt spontane Ca²⁺-Freisetzungen aus dem sarkoplasmatischen Retikulum, triggert verzögerte Nachdepolarisationen, verlängert das Aktionspotenzial und verstärkt die Heterogenität der Repolarisation, wodurch ST-Hebungen über veränderte transmurale Aktivierungsmuster zunehmen. Zusätzlich hält ein unter hypoxischen Bedingungen verstärkter später Na⁺-Strom (I_NaL) den Na⁺-Einstrom aufrecht, perpetuiert die Ca²⁺-Dysregulation und trägt zur elektrophysiologischen Grundlage von ST-Veränderungen bei.[20][26][27]
Die Entkopplung von Gap Junctions im hypoxischen Gewebe stört die interzelluläre Kommunikation und trägt ebenfalls zur ionischen Grundlage der ST-Hebung bei. Innerhalb von 10–15 Minuten nach Ischämiebeginn erhöhen intrazelluläres Ca²⁺ und Azidose den Gap-Junction-Widerstand, vor allem über Connexin-43-Kanäle, was zu einer elektrischen Isolation ischämischer Myozyten führt. Diese Entkopplung vermindert die Ausbreitung depolarisierender Ströme, reduziert die TQ-Senkung und moduliert die Amplitude der ST-Hebung durch heterogene Leitungsverzögerungen im Myokard. Die resultierenden Verletzungsströme verlaufen ungleichmäßig und verstärken dadurch die ST-Hebung als Ausdruck eines lokalisierten Leitungsblocks.[20][28]
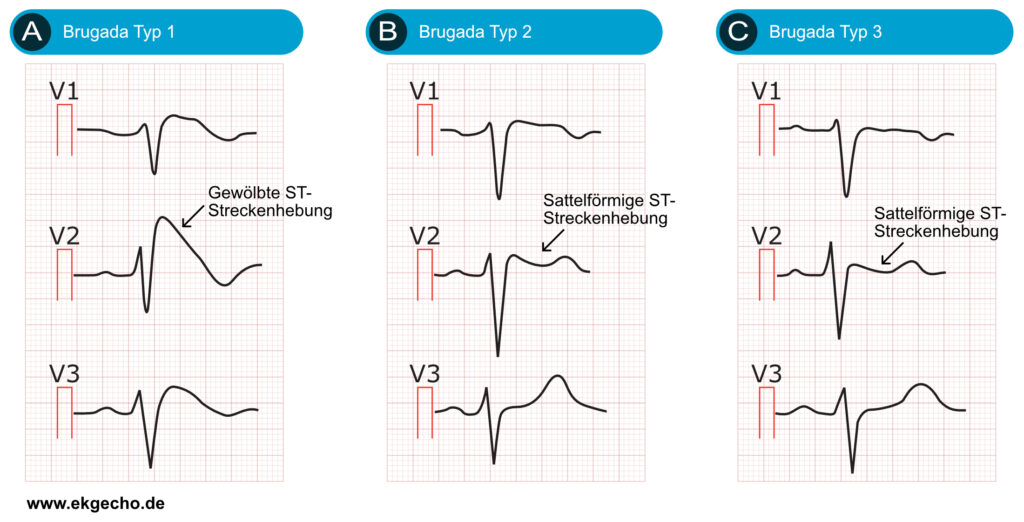
Genetische Faktoren, insbesondere Mutationen im SCN5A-Gen, das für die α-Untereinheit des kardialen spannungsabhängigen Na⁺-Kanals kodiert, bilden die molekulare Grundlage bestimmter Formen der ST-Hebung, vor allem beim Brugada-Syndrom. Funktionsverlustmutationen (z. B. Missense- oder Frameshift-Varianten) vermindern die I_Na-Dichte oder beschleunigen die Inaktivierung, sodass der transiente auswärtsgerichtete K⁺-Strom (I_to) im rechtsventrikulären Epikard ungebremst überwiegt. Dies führt zum Verlust des Aktionspotenzialdoms und erzeugt einen transmuralen Spannungsgradienten, der sich als sattelförmige oder gewölbte ST-Hebung in den präkordialen Ableitungen manifestiert und über einen Phase-2-Reentry zu Arrhythmien prädisponiert. Etwa 20–30 % der Brugada-Fälle sind mit SCN5A-Varianten assoziiert, was ihre Bedeutung bei hereditären Ionenkanalerkrankungen unterstreicht.[29][30]
Ätiologien
Ischämische Ursachen
Eine ST-Hebung ischämischer Genese ist am häufigsten Ausdruck eines ST-Hebungs-Myokardinfarkts (STEMI), einer Form des akuten Koronarsyndroms mit transmuraler Myokardischämie infolge eines vollständigen oder nahezu vollständigen Verschlusses einer Koronararterie, typischerweise nach Ruptur einer atherosklerotischen Plaque mit nachfolgender Thrombusbildung.[3] Dieser Verschluss führt zu persistierenden ST-Strecken-Hebungen im EKG und grenzt den STEMI vom Nicht-ST-Hebungs-Myokardinfarkt ab.[31] Das elektrokardiographische Kriterium für einen STEMI erfordert eine neu aufgetretene ST-Hebung am J-Punkt in mindestens zwei benachbarten Ableitungen mit Grenzwerten von ≥1 mm (0,1 mV) in allen Ableitungen außer V2–V3 sowie in V2–V3 von ≥2 mm (0,2 mV) bei Männern ≥40 Jahren, ≥2,5 mm (0,25 mV) bei Männern <40 Jahren und ≥1,5 mm (0,15 mV) bei Frauen.[32] Diese Veränderungen spiegeln transmurale Ischämiegradienten wider, bei denen Verletzungsströme vom ischämischen Epikard zum nichtischämischen Endokard die ST-Strecke verändern.[3]
Muster und Lokalisation der ST-Hebung liefern wichtige anatomische Hinweise auf die betroffene Koronararterie. Eine Vorderwandbeteiligung mit ST-Hebungen in V1–V4 spricht für einen Verschluss der proximalen oder mittleren linken Vorderwandarterie (LAD), die einen großen Teil des linken Ventrikels versorgt und aufgrund des potenziell ausgedehnten Myokardschadens prognostisch ungünstig ist.[33] Inferiore ST-Hebungen in II, III und aVF beruhen in etwa 80 % der Fälle auf einem Verschluss der rechten Koronararterie (RCA), seltener der linken Zirkumflexarterie (LCx), und können bei proximalem Verschluss mit einer Rechtsventrikelbeteiligung einhergehen.[33] Laterale Ischämien mit ST-Hebungen in I, aVL, V5 und V6 sind typischerweise mit einem LCx- oder Diagonalastverschluss assoziiert und können bis in posteriore Regionen reichen, die sich häufig nur indirekt über reziproke Veränderungen in anterioren Ableitungen erkennen lassen.[33]
Die EKG-Veränderungen beim STEMI entwickeln sich rasch nach arterieller Okklusion. Initial treten innerhalb von Sekunden bis Minuten hochspitze, symmetrische hyperakute T-Wellen als frühestes Ischämiezeichen auf, gefolgt von einer manifesten ST-Hebung innerhalb von 5–30 Minuten im Rahmen der entstehenden transmuralen Schädigung.[32] Die maximale Hebung wird innerhalb von Stunden erreicht; bei erfolgreicher Reperfusion kann sie sich zurückbilden, unbehandelt kommt es im Verlauf häufig zur Entwicklung pathologischer Q-Zacken und T-Negativierungen über Tage hinweg.[33]
Wesentliche Risikofaktoren des STEMI sind eine atherosklerotische Grunderkrankung, beschleunigt durch Rauchen, Diabetes mellitus, arterielle Hypertonie und Hyperlipidämie, die Plaqueinstabilität und Thrombose begünstigen.[3] In den USA ist die STEMI-Inzidenz infolge präventiver Maßnahmen zurückgegangen; für 2020 wurden etwa 162.000 Fälle pro Jahr (49 pro 100.000 Einwohner) angegeben, entsprechend ungefähr 20–30 % aller akuten Myokardinfarkte.[34]
Entzündliche und infektiöse Ursachen
Entzündliche und infektiöse Ursachen der ST-Hebung betreffen vor allem Perikard und Myokard und führen zu diffusen oder fokalen Repolarisationsstörungen im EKG. Die akute Perikarditis als häufige entzündliche Ursache manifestiert sich mit ausgedehnten konkaven ST-Strecken-Hebungen in mehreren Ableitungen – typischerweise I, II, aVL, aVF und V2–V6 – unter Aussparung von aVR und häufig auch V1 sowie mit PR-Streckensenkungen außerhalb von aVR.[35][36] Dieses Muster spiegelt eine subepikardiale Schädigung infolge der Perikardentzündung wider und unterscheidet sich von ischämischen Veränderungen durch seine nichtterritoriale Verteilung und das Fehlen reziproker ST-Senkungen.[37] Virale Infektionen, insbesondere durch Coxsackie-B-Viren, sind die häufigsten infektiösen Auslöser und machen bis zu 80–90 % der idiopathischen, vermutlich viralen Fälle aus; autoimmune Prozesse wie der systemische Lupus erythematodes sind in 5–15 % der Fälle beteiligt.[38][39] Ein wichtiger klinischer Hinweis ist ein pleuritischer Thoraxschmerz, der sich in Rückenlage verstärkt und beim Vorbeugen bessert, oft begleitet von einem auskultatorisch nachweisbaren Perikardreiben.[40]
Die Myokarditis, eine entzündliche Erkrankung des Myokards, häufig durch infektiöse Erreger ausgelöst, kann ST-Hebungen verursachen, die einen akuten Myokardinfarkt imitieren, meist jedoch diffuser oder fleckförmiger verteilt sind und keiner klaren Koronarversorgung folgen.[41] Virale Erreger, insbesondere Enteroviren einschließlich Coxsackie-B-Viren, sind für die Mehrzahl der Fälle verantwortlich; die entzündliche Zellinfiltration führt zu Myozytenschaden und Repolarisationsverschiebungen.[42] Im EKG können ST-Hebungen in inferioren und lateralen Ableitungen auftreten, oft begleitet von Arrhythmien wie ventrikulären Tachykardien oder AV-Blockierungen sowie unspezifischen T-Wellen-Veränderungen.[43] Kardiale Troponine sind aufgrund der Myokardschädigung häufig erhöht und helfen, die Myokarditis von einer isolierten Perikarditis abzugrenzen; die ST-Veränderungen bilden sich jedoch in bis zu 74 % der Fälle innerhalb von 24–48 Stunden zurück.[41] Anders als bei der Perikarditis ist der Schmerz meist weniger lageabhängig und klinisch schleichender; Echokardiographie oder kardiale MRT zeigen häufig Wandbewegungsstörungen oder Myokardödem und tragen zur Diagnosesicherung bei.[43]
Postkardiale Injury-Syndrome wie das Dressler-Syndrom, das 2–3 Wochen nach Myokardinfarkt auftreten kann, stellen eine autoimmun vermittelte Perikarditis mit rezidivierenden ST-Hebungen ähnlich der akuten Perikarditis dar.[44] Diese verzögerte Entzündungsreaktion geht häufig mit Perikard- und Pleuraergüssen einher, wird durch Antigene aus nekrotischem Myokard getriggert und zeigt in bis zu 60 % diffuse konkave ST-Hebungen und PR-Senkungen.[45] Das Syndrom ist im Reperfusionszeitalter seltener geworden, bleibt aber bei postinfarktbedingtem Fieber und Thoraxschmerz differenzialdiagnostisch relevant; erhöhte Entzündungsmarker wie C-reaktives Protein stützen die Diagnose.[44] Therapeutisch steht die antiinflammatorische Behandlung im Vordergrund; typischerweise normalisieren sich die EKG-Veränderungen zunächst und gehen anschließend in T-Negativierungen über.[45]
Nichtischämische und benigne Ursachen
Nichtischämische und benigne Ursachen der ST-Hebung umfassen eine Vielzahl metabolischer, struktureller, genetischer und physiologischer Zustände, die im EKG ischämische Muster vortäuschen können, ohne dass ein Koronarverschluss vorliegt. Diese Ätiologien beruhen auf Störungen des Elektrolythaushalts, strukturellen kardialen Anpassungen, Ionenkanaldysfunktionen oder extrakardialen Belastungen und erfordern häufig die Abgrenzung vom akuten Myokardinfarkt anhand des klinischen Kontextes und ergänzender Diagnostik. Das Verständnis dieser Muster ist entscheidend, um unnötige Interventionen zu vermeiden.
Elektrolytstörungen, insbesondere die Hyperkaliämie, können durch Veränderungen der myokardialen Repolarisation Pseudo-ST-Hebungen verursachen. Bei schwerer Hyperkaliämie, typischerweise bei Serumkaliumwerten über 7 mEq/L, zeigen sich im EKG spitze T-Wellen, die in ein Pseudo-Infarktmuster mit ST-Hebung übergehen können, meist infolge einer beschleunigten Repolarisation und begleitender Leitungsstörungen. Diese Pseudo-Hebung beruht auf einer globalen Verkürzung der Aktionspotenzialdauer und einer dadurch synchroneren ventrikulären Repolarisation und kann einen STEMI vortäuschen, wie es etwa bei akuter Nierenschädigung beobachtet wird. Die rasche Erkennung ist essenziell, da sich diese Veränderungen nach Kaliumkorrektur zurückbilden.
Strukturelle kardiale Veränderungen wie die linksventrikuläre Hypertrophie (LVH) und Schenkelblöcke verursachen häufig sekundäre ST-Hebungen infolge nichtischämischer Repolarisationsstörungen. Bei LVH, meist Folge einer chronischen Druckbelastung wie arterieller Hypertonie, zeigt das Strain-Muster typischerweise ST-Senkungen und T-Negativierungen in den lateralen Ableitungen (I, aVL, V5–V6), während in den rechtspräkordialen Ableitungen (V1–V3) reziproke ST-Hebungen infolge veränderter ventrikulärer Aktivierung und hypertrophiebedingter Repolarisationsgradienten auftreten können. Persistierende ST-Hebungen bei LVH spiegeln diese chronischen Adaptationen wider und müssen von akuten Läsionen unterschieden werden. Ähnlich sind beim Linksschenkelblock (LBBB) diskordante ST-Hebungen häufig ein benigner Begleitbefund; zur Identifikation ischämischer Äquivalente dienen die Sgarbossa-Kriterien: konkordante ST-Hebung ≥1 mm (Score 5), konkordante ST-Senkung ≥1 mm in V1–V3 (Score 3) oder exzessiv diskordante ST-Hebung ≥5 mm (Score 2), wobei ein Score ≥3 auf eine akute Koronarokklusion hinweist. Diese in mehreren Studien validierten Kriterien betonen die proportionale Beurteilung, um eine Überdiagnose von Ischämie bei LBBB zu vermeiden.
Genetische Kanalopathien, exemplarisch das Brugada-Syndrom, zeigen charakteristische ST-Hebungsmuster mit erhöhter Gefahr des plötzlichen Herztodes. Das Brugada-Syndrom Typ 1 ist durch eine gewölbte ST-Hebung ≥2 mm in V1–V3 mit anschließender negativer T-Welle gekennzeichnet. Ursache ist eine Natriumkanaldysfunktion (SCN5A-Mutationen), die einen transmuralen Spannungsgradienten im rechtsventrikulären Ausflusstrakt erzeugt. Dieses diagnostische EKG-Muster, spontan oder nach Provokation mit Natriumkanalblockern, identifiziert Patienten mit hohem Risiko ventrikulärer Arrhythmien; bei symptomatischen Betroffenen muss eine ICD-Implantation erwogen werden.
Weitere nichtischämische Ursachen sind die Lungenembolie (LE) und neurogene Effekte bei intrakranieller Blutung. Bei akuter LE kann eine rechtsventrikuläre Belastung durch akute Druckerhöhung zu ST-Hebungen in den rechtspräkordialen Ableitungen (V1–V3) oder in der rechten Armableitung führen, oft zusammen mit einem S1Q3T3-Muster und T-Negativierungen, als Ausdruck einer pulmonalen Hypertonie ohne koronare Ursache. Rechtsseitige EKG-Ableitungen können qr- oder qs-Muster mit ST-Hebung zeigen und mit einer ungünstigen Prognose korrelieren. Neurogene ST-Hebungen treten bei intrakraniellen Blutungen, insbesondere bei Subarachnoidalblutung, infolge einer Katecholaminexzesse mit myokardialem Stunning auf; EKG-Veränderungen mit STEMI-ähnlicher ST-Hebung finden sich in bis zu 25 % der Fälle, zusätzlich häufig QT-Verlängerung und T-Negativierungen. Diese Veränderungen werden durch autonome Dysregulation und neurokardiogene Schädigung vermittelt, bilden sich aber meist unter Behandlung der Blutung zurück; dennoch muss eine kardiale Ischämie ausgeschlossen werden.
Benigne physiologische Varianten wie die Frührepolarisation bei Athleten und ethnische Unterschiede können anteriore ST-Hebungen ohne pathologische Bedeutung verursachen. Bei Sportlern, insbesondere jungen Männern, zeigt sich die benigne Frührepolarisation mit einer J-Punkt-Hebung ≥1 mm und konkav ansteigender ST-Strecke in den präkordialen Ableitungen als Ausdruck von Vagotonus und sportbedingtem kardialem Remodeling; die Prävalenz liegt bei trainierten Personen bei bis zu 13 %. Dieses Muster ist bei schwarzen Athleten häufiger; ethnische Varianten umfassen höhere Raten anteriorer ST-Hebungen (bis zu 20–30 %) und inferolateraler J-Wellen, häufig assoziiert mit erhöhter linksventrikulärer Masse, jedoch mit geringem Arrhythmierisiko, solange die Veränderungen auf V2–V4 begrenzt sind und keine Symptome vorliegen. Diese Befunde unterstreichen die Notwendigkeit populationsspezifischer EKG-Normen im Screening, um Fehldiagnosen zu vermeiden.
Klinische Beurteilung
Diagnostische EKG-Kriterien
Die ST-Strecken-Hebung im Elektrokardiogramm (EKG) ist ein zentrales diagnostisches Merkmal des akuten Myokardinfarkts, insbesondere des ST-Hebungs-Myokardinfarkts (STEMI). Nach der ACC/AHA-Leitlinie 2025 erfordert die Diagnose eine neu aufgetretene ST-Hebung am J-Punkt in mindestens zwei benachbarten Ableitungen, mit Schwellenwerten von ≥1 mm (0,1 mV) in allen Ableitungen außer V2–V3 (einschließlich der Extremitätenableitungen I, II, III, aVR, aVL, aVF sowie der Brustwandableitungen V4–V6). Für V2–V3 gelten geschlechts- und altersspezifische Grenzwerte: ≥2 mm (0,2 mV) bei Männern ≥40 Jahren, ≥2,5 mm (0,25 mV) bei Männern <40 Jahren und ≥1,5 mm (0,15 mV) bei Frauen. Diese Kriterien entsprechen weitgehend den ESC-Leitlinien 2023, die ebenfalls ≥1 mm in anderen benachbarten Ableitungen und vergleichbare Schwellenwerte für V2–V3 festlegen und betonen, dass die Hebung diese Werte erreichen oder überschreiten muss, um diagnostisch relevant zu sein.[2][46]
Benachbarte Ableitungen sind anatomisch angrenzende Ableitungen im Standard-12-Kanal-EKG, beispielsweise I und aVL (lateral), II, III und aVF (inferior) oder V1 bis V6 (anterior bzw. septal). Die Hebung muss in mindestens zwei solchen Ableitungen nachweisbar sein, um ein regionales Ischämiegebiet anzuzeigen und Artefakte oder isolierte Normalvarianten weniger wahrscheinlich zu machen. Diese Anforderung stellt sicher, dass das Muster eher einer Koronarversorgung als Rauschen oder einer physiologischen Variante entspricht.[2][46]
Die Morphologie der ST-Strecke liefert zusätzlichen diagnostischen Kontext. Konvexe („nach oben gewölbte“) oder gerade/schräge Morphologien sprechen stark für eine ischämische Schädigung, wie beim STEMI, bei dem die Hebung häufig in eine prominente T-Welle übergeht. Konkave, aufsteigende Verläufe sind demgegenüber eher typisch für eine benigne Frührepolarisation oder eine Perikarditis, während schräg ansteigende Verläufe auch bei linksventrikulärer Hypertrophie ohne akute Ischämie vorkommen können. Diese Muster unterstützen die Differenzierung pathologischer von physiologischen Hebungen, sind für sich genommen jedoch nicht beweisend und müssen stets im Zusammenhang mit Grenzwerten und klinischem Kontext interpretiert werden.[2][4]
Serielle EKGs sind essenziell, um dynamische Veränderungen zu bestätigen, da sich ST-Hebungen im Zeitverlauf verändern können. Nach Reperfusion weist eine erfolgreiche Wiedereröffnung des Gefäßes typischerweise innerhalb von 90 Minuten zu einer Rückbildung der ST-Hebung um ≥50 % in anterioren Ableitungen bzw. ≥70 % in inferioren Ableitungen; ein Persistieren unterhalb dieser Schwellen spricht für ein Reperfusionsversagen und eine ungünstigere Prognose. Initiale und Verlaufsekgs sollten idealerweise innerhalb von 10 Minuten nach Erstkontakt abgeleitet und bei Bedarf wiederholt werden, um Progression oder Rückbildung zu dokumentieren und dringliche Interventionen zu steuern. Die Messung der ST-Strecke erfolgt am J-Punkt unter Bezug auf die PR-Strecke als Grundlinie.[2][46]
Strategien der Differenzialdiagnose
Die Differenzialdiagnose der ST-Hebung stützt sich auf die Integration von Anamnese und klinischer Untersuchung, um die nachfolgende Diagnostik zielgerichtet zu lenken und die wahrscheinlichen Ursachen einzugrenzen. Patienten mit ischämischer Genese, insbesondere STEMI, berichten häufig über drückende retrosternale Schmerzen mit Ausstrahlung in Arm, Kiefer oder Rücken, Dauer über 20 Minuten und fehlender Besserung in Ruhe oder nach Nitroglyzerin.[47] Demgegenüber präsentiert sich die Perikarditis typischerweise mit pleuritischem Thoraxschmerz, der sich in Rückenlage verstärkt und im Sitzen mit Vorbeugen bessert.[47] Eine kürzlich durchgemachte Virusinfektion oder Fieber spricht für entzündliche oder infektiöse Ursachen wie Perikarditis oder Myokarditis.[40] In der körperlichen Untersuchung spricht ein Perikardreiben – ein hochfrequentes, kratzendes Geräusch, am besten links parasternal hörbar – stark für eine Perikarditis, während Zeichen der Herzinsuffizienz wie Halsvenenstauung oder pulmonale Rasselgeräusche auf eine myokardiale Ischämie oder einen Infarkt hinweisen können.[40]
Die Labordiagnostik ergänzt die Anamnese, indem sie Marker der Myokardschädigung, der Entzündung oder metabolischer Störungen erfasst. Kardiale Troponine sind sowohl beim STEMI als auch bei der Myokarditis infolge von Myozytenschädigung erhöht; bei der Myokarditis sind die Anstiege häufig geringer und persistieren länger, ohne das für den akuten Infarkt typische rasche An- und Abfluten. Serielle Bestimmungen im Abstand von 3–6 Stunden erleichtern die Differenzierung.[48] Entzündungsmarker wie C-reaktives Protein (CRP) und Blutsenkungsgeschwindigkeit (BSG) sind bei Perikarditis und Myokarditis meist erhöht; ein CRP >3 mg/L in akuten Fällen stützt die Diagnose einer nichtischämischen Entzündung.[40] Elektrolytbestimmungen sollten insbesondere auf Störungen wie eine Hyperkaliämie (Serumkalium >5,5 mEq/L) prüfen, die Pseudo-ST-Hebungen mit STEMI-Mimik verursachen kann, vor allem bei Patienten mit Nierenerkrankung oder entsprechender Medikation.[49]
Bildgebende Verfahren liefern strukturelle und funktionelle Zusatzinformationen. Die transthorakale Echokardiographie ist ein Verfahren der ersten Linie zum Nachweis regionaler Wandbewegungsstörungen, etwa einer Hypokinesie im Versorgungsgebiet einer verschlossenen Koronararterie beim STEMI, im Gegensatz zu einer eher diffusen Hypokontraktilität bei Myokarditis.[50] Eine CT-Pulmonalisangiographie ist indiziert, wenn der Verdacht auf Lungenembolie besteht; sie zeigt Füllungsdefekte in den Pulmonalarterien und eine rechtsventrikuläre Belastung als mögliche Erklärung der EKG-Veränderungen.[51] Die kardiale Magnetresonanztomographie bietet bei Verdacht auf Myokarditis eine hohe Spezifität durch Late-Gadolinium-Enhancement-Muster und Ödemnachweis, insbesondere wenn die Echokardiographie keine eindeutigen Befunde liefert.[52]
Spezifische Scores unterstützen in ausgewählten Situationen die Risikostratifizierung und diagnostische Präzision. Bei Patienten mit Linksschenkelblock, der die Beurteilung von ST-Hebungen erschwert, verbessern die modifizierten Sgarbossa-Kriterien die Sensitivität für den Nachweis eines akuten Myokardinfarkts: konkordante ST-Hebung ≥1 mm (5 Punkte), konkordante ST-Senkung ≥1 mm in V1–V3 (3 Punkte) oder exzessiv diskordante ST-Hebung ≥25 % der vorausgehenden S-Wellen-Tiefe (2 Punkte), wobei ein Gesamtscore ≥3 eine hohe Wahrscheinlichkeit einer akuten Myokardischämie anzeigt. Für akute Koronarsyndrome einschließlich STEMI-Äquivalenten nutzt der TIMI-Risikoscore sieben Variablen – darunter Alter ≥65 Jahre, ≥3 kardiale Risikofaktoren und ST-Abweichung ≥0,5 mm – zur Abschätzung der 30-Tage-Mortalität; Werte ≥3 identifizieren Patienten, die besonders von einer aggressiven Reperfusionsstrategie profitieren.[53] In Kombination mit EKG-Merkmalen wie konkaver versus konvexer Morphologie ermöglicht dies ein systematisches Vorgehen zur Abgrenzung benigner von vital bedrohlichen Ursachen.[54]
Management und Outcomes
Akute Interventionen
Das akute Management einer ST-Hebung erfordert zunächst die rasche Zuordnung der zugrunde liegenden Ursache, da Verzögerungen mit schweren Folgen wie Arrhythmien oder hämodynamischer Instabilität verbunden sein können.[2] Initiale supportive Maßnahmen – einschließlich Sauerstoffgabe bei Hypoxämie (Sauerstoffsättigung <90 %) und Betablockade zur Frequenzkontrolle ohne Kontraindikationen, z. B. niedrig dosiertes orales Metoprolol innerhalb von 24 Stunden bei STEMI oder nichtischämischen ST-Hebungen, jedoch nicht bei akuter Herzinsuffizienz, Bradykardie oder Hypotonie – werden mit ätiologiespezifischen Therapien kombiniert, um den Patienten zu stabilisieren. Nitroglyzerin kann sublingual oder intravenös bei persistierender Ischämie oder Hypertonie im Rahmen eines STEMI eingesetzt werden; ein routinemäßiger Morphineinsatz wird wegen potenzieller Risiken zurückhaltend beurteilt.[2]
Beim ST-Hebungs-Myokardinfarkt (STEMI) ist die unverzügliche Reperfusionstherapie zusammen mit einer antithrombotischen Behandlung der zentrale Baustein. Acetylsalicylsäure wird so früh wie möglich in einer Aufsättigungsdosis von 162–325 mg oral (nicht magensaftresistent) verabreicht, gefolgt von 75–100 mg täglich als Erhaltungstherapie (Klasse I, Evidenzgrad A).[2] Ein P2Y12-Inhibitor wie Ticagrelor (180 mg Aufsättigung) oder Prasugrel (60 mg Aufsättigung) ist bei Patienten mit geplanter perkutaner Koronarintervention (PCI) Clopidogrel vorzuziehen; die duale Plättchenhemmung sollte mindestens 12 Monate fortgeführt werden (Klasse I, Evidenzgrad A/B-R).[2] Eine parenterale Antikoagulation wird als Begleittherapie empfohlen (Klasse I, Evidenzgrad A); bei primärer PCI wird intravenöses Enoxaparin gegenüber unfraktioniertem Heparin bevorzugt (Enoxaparin: 30 mg i.v. Bolus plus 1 mg/kg s.c. alle 12 Stunden; Alternative UFH: initial 60 U/kg [max. 4000 U], anschließend 12 U/kg/h [max. 1000 U/h], mit Steuerung über die aktivierte Gerinnungszeit während der PCI).[2] Die primäre PCI ist die bevorzugte Reperfusionsstrategie; angestrebt wird eine Door-to-Balloon-Zeit von ≤90 Minuten bei Vorstellung in einem PCI-fähigen Zentrum. Wenn die erwartete Zeit vom ersten medizinischen Kontakt bis zur Drahtpassage 120 Minuten überschreitet, ist eine Fibrinolyse (z. B. Tenecteplase oder Alteplase) mit einer Door-to-Needle-Zeit ≤30 Minuten indiziert, gefolgt von einer Koronarangiographie innerhalb von 3–24 Stunden (Klasse I, Evidenzgrad A/B-R).[2]
Bei akuter Perikarditis mit diffuser ST-Hebung wird rasch eine antiinflammatorische Therapie eingeleitet, um Schmerzen und Entzündung zu kontrollieren und gleichzeitig Substanzen mit erhöhtem Blutungsrisiko zu vermeiden. Nichtsteroidale Antirheumatika (NSAR) wie Ibuprofen (600–800 mg alle 8 Stunden) oder Acetylsalicylsäure (750–1000 mg alle 8 Stunden) gelten für 1–2 Wochen als Erstlinientherapie und werden entsprechend dem klinischen Ansprechen ausgeschlichen; eine gastroprotektive Begleittherapie mit Protonenpumpenhemmern wird empfohlen (Klasse I, Evidenzgrad A).[55] Colchicin wird zusätzlich als Erstlinientherapie in einer Dosierung von 0,5 mg einmal oder zweimal täglich – angepasst an das Körpergewicht – über 3 Monate empfohlen, um das Rezidivrisiko zu senken (Klasse I, Evidenzgrad A).[55] Bei rezidivierender oder refraktärer Perikarditis trotz NSAR, Colchicin und Kortikosteroiden werden Anti-IL-1-Therapien wie Anakinra oder Rilonacept empfohlen (Klasse I, Evidenzgrad B).[56] Antikoagulanzien sind wegen des erhöhten Risikos eines Hämoperikards kontraindiziert (Klasse III, Evidenzgrad C).[55]
Bei hyperkaliämiebedingter ST-Hebung, die im EKG einen STEMI vortäuschen kann, sind eine sofortige Membranstabilisierung und eine rasche intrazelluläre Kaliumverschiebung entscheidend, um lebensbedrohliche Arrhythmien zu verhindern. Initial wird Calciumglukonat intravenös (10 mL einer 10%igen Lösung über 2–5 Minuten) oder alternativ Calciumchlorid verabreicht, um die kardialen Membraneffekte zu antagonisieren; der Wirkungseintritt erfolgt nach 1–3 Minuten, die Wirkdauer beträgt 30–60 Minuten, bei persistierenden EKG-Veränderungen kann eine Wiederholung erforderlich sein (Klasse I, Evidenzgrad B).[57] Parallel erfolgt eine Insulin-Glukose-Therapie (10 IE Normalinsulin zusammen mit 25–50 g Glukose i.v.), um Kalium intrazellulär zu verschieben; der Wirkungseintritt liegt bei 30–60 Minuten, wobei auf Hypoglykämien zu achten ist (Klasse I, Evidenzgrad A).[57] Die Serumkaliumwerte sollten innerhalb von 60 Minuten erneut kontrolliert werden, um weitere eliminierende Maßnahmen zu steuern.[57]
Prognostische Faktoren
Bei Patienten mit ST-Hebungs-Myokardinfarkt (STEMI) liegt die Krankenhausmortalität bei zeitgerechter Reperfusionstherapie typischerweise zwischen 5 % und 10 %; in aktuellen Registern werden für die primäre perkutane Koronarintervention (PCI) Raten von etwa 4–9 % berichtet.[58][59] Wesentliche ungünstige prognostische Faktoren sind ein höheres Alter über 75 Jahre, das in älteren Kollektiven mit Mortalitätsraten von über 20–25 % assoziiert ist, eine Vorderwandlokalisation mit größerem Infarktareal sowie eine verzögerte PCI. Systemverzögerungen von mehr als 2 Stunden gehen mit einer schrittweise zunehmenden 30-Tage-Mortalität einher, mit einem relativen Anstieg um ungefähr 7,5 % pro 30 Minuten Verzögerung bzw. etwa 1 % absolut pro Stunde.[60][61][62]
Bei benignen Ursachen der ST-Hebung, etwa Frührepolarisationsmustern bei asymptomatischen Personen, ist die Prognose exzellent; es besteht kein erhöhtes Risiko unerwünschter kardialer Ereignisse, und über Aufklärung sowie gegebenenfalls Verlaufskontrollen hinaus ist keine spezifische Therapie erforderlich.[63][64] Diese Muster finden sich häufig bei jungen, gesunden Männern und bilden sich meist im Verlauf ohne Folgeschäden zurück.[12]
Zu den häufigen Komplikationen in Konstellationen mit ST-Hebung, insbesondere beim STEMI, zählen lebensbedrohliche Arrhythmien wie ventrikuläre Tachykardien oder Kammerflimmern, die in bis zu 10–15 % der Fälle auftreten und wesentlich zur Frühmortalität beitragen, sowie eine akute Herzinsuffizienz, die sich bei etwa 20–30 % der Patienten infolge einer Pumpfunktionsstörung entwickelt und eine führende Ursache des intrahospitalen Versterbens darstellt.[65][66] Der GRACE-Risikoscore integriert Parameter wie Alter, Herzfrequenz, systolischen Blutdruck, Kreatinin, Killip-Klasse, Herzstillstand, ST-Strecken-Abweichung und Enzymwerte zur Prognoseabschätzung bei akuten Koronarsyndromen einschließlich STEMI. Er sagt die 6-Monats-Mortalität mit hoher Genauigkeit (c-Statistik >0,80) voraus und hilft, die Intensität der Versorgung zu steuern.[67][68]
Die Langzeitprognose nach Ereignissen mit ST-Hebung wird wesentlich durch Sekundärprävention beeinflusst. Statine senken durch Lipidreduktion rezidivierende ischämische Ereignisse um 20–30 %, und ACE-Hemmer reduzieren die Mortalität um 15–20 % durch Nachlastsenkung und Hemmung des ventrikulären Remodelings; beide sollten bei geeigneten Patienten in der Regel langfristig fortgeführt werden.[69][70] Bei nichtischämischen Ursachen wie der Myokarditis liegt das 1-Jahres-Überleben bei etwa 80–90 %; die meisten Patienten erholen sich vollständig, während eine persistierende ventrikuläre Dysfunktion mit einem höheren Risiko einhergeht.[71][72]
Literaturverzeichnis
ST Segment – StatPearls – NCBI Bookshelf – NIH
ACC/AHA Guidelines for the Management of Patients With ST …
Acute ST-Segment Elevation Myocardial Infarction (STEMI) – NCBI
ST elevation: Differential diagnosis and caveats. A comprehensive …
ST-Segment Elevation: Defined by the Company It Keeps – PMC
The ST Segment – ECG Library Basics – LITFL
ECG in myocardial ischemia: ischemic changes in the ST segment …
AHA/ACCF/HRS Recommendations for the Standardization and …
Naming of the Waves in the ECG, With a Brief Account of Their …
Characteristics of the normal ECG (P-wave, QRS complex, ST …
Benign Early Repolarisation – ECG Library Diagnosis – LITFL
Electrocardiographic Early Repolarization | Circulation
Early Repolarization vs. Acute Pericarditis Morphology – NIH
The Phenomenon of Early Repolarization | Circulation
Benign Early Repolarization/Subtle ST Elevation in a Young Patient …
Does early repolarization on ECG increase the risk of cardiac death …
ECG Cases 2: Early Repolarization or Anterior STEMI?
Acute Myocardial Ischemia: Cellular Mechanisms Underlying ST …
Acute myocardial ischemia: cellular mechanisms underlying ST …
Cellular basis for ST-segment changes observed during ischemia
Mechanism of S-T Segment Alteration During Acute Myocardial Injury
Mechanism and time course of S-T and T-Q segment … – PubMed
The ST injury vector: electrocardiogram-based estimation of location …
Late sodium current and calcium homeostasis in arrhythmogenesis
Changes in Myocardial Electrical Impedance Induced by Coronary …
Cellular Basis for the Brugada Syndrome and Other Mechanisms of …
Brugada syndrome: A comprehensive review of pathophysiological …
ECG Diagnosis: ST-Elevation Myocardial Infarction – PMC – NIH
AHA/ACCF/HRS Recommendations for the Standardization and …
The electrocardiogram in ST elevation acute myocardial infarction
Cover Story | Acute Coronary Syndromes: New Perspectives, New …
Management of Acute and Recurrent Pericarditis – JACC
Pericardial Disease | Circulation
Electrocardiographic Manifestations and Differential Diagnosis of …
Pericarditis – StatPearls – NCBI Bookshelf – NIH
Acute Pericarditis: Rapid Evidence Review – AAFP
Diagnosis of acute pericarditis – European Society of Cardiology
Diagnostic and prognostic role of electrocardiogram in acute … – NIH
Viral Myocarditis | Circulation – American Heart Association Journals
Review Acute myocarditis: aetiology, diagnosis and management
Post-cardiac injury syndrome: aetiology, diagnosis, and treatment
Dressler’s syndrome: are we underdiagnosing what we think to … – NIH
2025 ACC/AHA/ACEP/NAEMSP/SCAI Guideline for the …
2023 ESC Guidelines for the management of acute coronary …
Acute Pericarditis Differential Diagnoses – Medscape Reference
Differential diagnosis of elevated troponins – PMC – NIH
ST-Segment Elevation Resulting From Hyperkalemia | Circulation
Myocardial infarction | Radiology Reference Article | Radiopaedia.org
Pulmonary Emboli Mimicking ST-Elevation Myocardial Infarction …
Acute Myocarditis: Noninvasive Evaluation with Cardiac MRI and …
Application of the TIMI Risk Score for ST-Elevation MI in the National …
ST-segment elevation: Differential diagnosis, caveats
[PDF] Acute hyperkalemia in the emergency department – KDIGO
Reperfusion therapies and in-hospital outcomes for ST-elevation …
Factors of Hospital Mortality in Men and Women with ST-Elevation …
STEMI-OP in-hospital mortality prediction algorithms – Nature
The influence of system delay on 30-day and on long-term mortality …
System Delay and Mortality Among Patients With STEMI Treated …
Early Repolarisation – What Should the Clinician Do? | AER Journal
Complications of Myocardial Infarction – Medscape Reference
STEMI: ST-segment Elevation Myocardial Infarction
GRACE ACS Risk and Mortality Calculator – MDCalc
In-Hospital Prognostic Stratification of STEMI Patients Using the …
Secondary prevention following myocardial infarction: a clinical update
Pharmacological secondary prevention of MI – Beggs – 2021
Myocarditis: Symptoms and Causes – Cleveland Clinic
Trends in myocarditis incidence, complications and mortality in …