
QT-Zeit-Verlängerung, Long-QT-Syndrom (LQTS) und Torsades-de-Pointes-Tachykardie
QT-Zeit-Verlängerung, Long-QT-Syndrom (LQTS) und Torsades-de-Pointes-Tachykardie
Die QT-Zeit ist das Zeitintervall vom Beginn des QRS-Komplexes bis zum Ende der T-Welle. Dieses Intervall verkörpert die Gesamtzeit, die zur De- und Repolarisierung der Ventrikel erforderlich ist (Abbildung 1). Die Länge der QT-Zeit korreliert stark mit dem Risiko für potenziell lebensbedrohliche ventrikuläre Arrhythmien. Daher muss die QT-Zeit bei der Befundung eines EKGs immer beurteilt werden. Das Long-QT-Syndrom (LQTS) manifestiert sich, wenn eine langes QT-Zeit ventrikuläre Arrhythmien induziert.

Die QT-Zeit hängt umgekehrt von der Herzfrequenz ab. Wenn die Herzfrequenz steigt, nimmt die QT-Zeit ab und umgekehrt. Um zu beurteilen, ob die QT-Zeit normal ist oder nicht, muss man daher die aktuelle Herzfrequenz berücksichtigen. Dies geschieht durch eine Anpassung der QT-Zeit an die Herzfrequenz, und die resultierende angepasste QT-Zeit wird als korrigierte QT-Zeit oder einfach QTc-Zeit bezeichnet. Die primäre Gefahr liegt in langen QTc-Intervallen, da sie für eine sehr instabile polymorphe ventrikuläre Tachykardie prädisponieren, die Torsade-de-Pointes-Tachykardie genannt wird. Eine ungewöhnlich kurze QTc-Zeit ist ebenfalls arrhythmogen, aber dies ist eine sehr seltene Erkrankung.
Viele Formeln wurden vorgeschlagen, um korrigierte QT-Intervalle zu berechnen. Im Folgenden werden einige dieser Formeln vorgestellt:

Bazett-Formel: QTc = QT-Intervall / √ (RR-Intervall)
Fridericia-Formel: QTc = QT-Intervall / (RR-Intervall)1/3
Framingham-Formel: QTc = QT-Intervall + 154 x (1 – RR-Intervall)
Hodges-Formel: QTc = QT-Intervall + 1.75 x [(60 / RR-Intervall) − 60]
RR-Intervall = 60 / HR
Die Bazett-Formel ist die am häufigsten verwendete Formel. Alle oben aufgeführten Formeln wurden jedoch vor mehreren Jahrzehnten entwickelt und haben Nachteile, die sie für die klinische Praxis ungeeignet machen. Stattdessen wird empfohlen, die automatische (maschinelle) Berechnung der korrigierten QT-Zeit zu verwenden. Diese QTc wird in allen modernen EKG-Geräten berechnet und die verwendeten Formeln sind präziser als die oben aufgeführten. Es kann weiterhin empfohlen werden, eine QTc-Zeit-Verlängerung manuell zu überprüfen. Die manuelle Messung wird durchgeführt, indem das Intervall zwischen dem ersten (frühesten) Zeichen der ventrikulären Depolarisation (in einer beliebigen Ableitung) bis zum letzten Anzeichen einer ventrikulären Repolarisation (in einer beliebigen Ableitung) gemessen wird.
QT-Zeit-Verlängerung verursacht Long-QT-Syndrom
Eine ungewöhnlich verlängerte QTc-Zeit wird als QTc-Zeit-Verlängerung (oder nur QT-Zeit-Verlängerung ohne „c“) bezeichnet. Die obere Referenzgrenze für die QTc-Zeit beträgt bei Männern 460 ms und bei Frauen 470 ms. Eine QTc-Zeit, die diese Grenzwerte überschreitet, kann eine Torsade-de-Pointes-Tachykardie verursachen. Wenn dies der Fall ist, d.h. wenn eine Person mit einer QT-Zeit-Verlängerung solche ventrikulären Arrhythmien entwickelt, wird diese Erkrankung als Long-QT-Syndrom (LQTS) bezeichnet.
Ursachen für eine QT-Zeit-Verlängerung
Eine QT-Zeit-Verlängerung ist entweder angeboren (genetisch) oder erworben.
Das kongenitale Long-QT-Syndrom wird durch Mutationen in kardialen Ionenkanälen verursacht. Es wurden mehr als 10 Arten von kongenitaler QT-Zeit-Verlängerung entdeckt. Die angeborene QT-Zeit-Verlängerung ist eine sehr ernste Erkrankung mit hoher Mortalität. Bei unbehandelten Patienten, die eine Synkope erlebt haben, sterben 20 % innerhalb eines Jahres. Glücklicherweise kann diese Sterblichkeitszahl durch eine evidenzbasierte Therapie über einen Follow-Up-Zeitraum von 15 Jahren auf 1 % gesenkt werden. Drei Arten von LQTS (LQT1, LQT2 und LQT3) machen etwa 90 % aller Fälle von angeborenem LQTS aus. Es wird geschätzt, dass die Prävalenz der angeborenen QT-Zeit-Verlängerung 1 pro 2000 Individuen in der Bevölkerung beträgt (Prävalenzzahlen aus Italien). Wichtig ist, dass Personen mit angeborener QT-Zeit-Verlängerung häufig berichten, dass unerklärliche Synkopen oder plötzlicher Herzstillstand in der Familie aufgetreten seien. Eine solche Familienanamnese ist ein starker Indikator für den plötzlichen Herztod.
Das erworbene Long-QT-Syndrom wird durch Medikamente (Amiodaron, Sotalol, Procainamid), Hypokaliämie, Hypomagnesiämie und ausgeprägte Bradykardie verursacht. Da jeder dieser Faktoren (Medikamente, Elektrolytstörungen und Bradykardie) durchaus häufig auftritt, aber nur bei einigen Individuen eine QT-Zeit-Verlängerung zu verursachen scheint, wird angenommen, dass es eine zugrunde liegende genetische Anfälligkeit für die Entwicklung eines erworbenen Long-QT-Syndroms geben muss.
Das Risiko, eine Torsade-de-Pointes-Tachykardie (polymorphe ventrikuläre Tachykardie) zu entwickeln, ist sowohl bei angeborenen als auch bei erworbener QT-Zeit-Verlängerung offensichtlich. Je länger die QT-Zeit ist, desto größer ist das Risiko, eine Torsade-de-Pointes-Tachykardie zu entwickeln. Im Allgemeinen entwickelt sich eine Torsade-de-Pointes-Tachykardie bei QTc-Intervallen von mehr als 490 Millisekunden.
Eine Torsade-de-Pointes-Tachykardie wird normalerweise durch eine ventrikuläre Extrasystole induziert, die früh im Herzzyklus auftritt. Das Risiko für eine Torsade-de-Pointes-Tachykardie steigt unter Bradykardie. Eine Torsade-de-Pointes-Tachykardie verursacht eine Synkope (oder Präsynkope), aber normalerweise ist die Arrhythmie selbstlimitierend (innerhalb von 30 Sekunden). Eine Minderheit von Fällen von Torsade-de-Pointes-Tachykardie geht in Kammerflimmern über, was ohne sofortige Behandlung tödlich verläuft. Abbildung 2 zeigt eine Torsade-de-Pointes-Tachykardie.

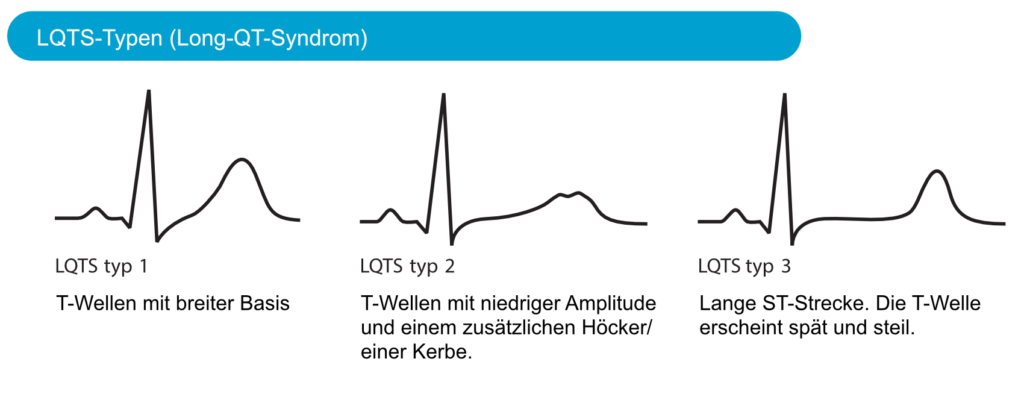
Neben der QT-Zeit selbst kann die T-Welle wertvolle Informationen über die Art des Long-QT-Syndroms liefern; insbesondere kann sie zwischen Typ 1 LQTS, Typ 2 LQTS und Typ 3 LQTS unterscheiden. Die T-Welle sollte in den Brustwandableitungen beurteilt werden. Siehe Abbildung 3. Gelegentlich zeigen Personen mit LQTS eine alternierende T-Welle, was bedeutet, dass die Amplitude oder Richtung der T-Welle von einem Schlag zum nächsten wechselt. Eine alternierende T-Welle ist ein Zeichen für ein sehr hohes Risiko für Torsade-de-Pointes-Tachykardien. Ebenso können Sinuspausen bei angeborenem LQTS auftreten.


EKG-Kriterien der Torsade-de-Pointes-Tachykardie
- QTc-Zeit-Verlängerung vor dem Beginn der Torsade-de-Pointes-Tachykardie.
- Spindelförmiges Oszillieren der QRS-Komplexe um die isoelektrische Linie (polymorphe ventrikuläre Tachykardie).
Kongenitales Long-QT-Syndrom (LQTS)
Mindestens 13 Varianten von kongenitalem LQTS wurden beschrieben. Die Mutationen sind durch eine autosomale Vererbung mit reduzierter Penetranz gekennzeichnet. LQTS Typ 1, Typ 2 und Typ 3 (genannt LQT1, LQT2 und LQT3) machen 90 % aller Fälle von Long-QT-Syndrom aus. LQT1 und LQT2 repräsentieren jeweils etwa 40 % aller Fälle.
Typ 1 des Long-QT-Syndroms (LQT1) wird durch eine Mutation im Kaliumkanal KCNQ1 (Funktionsverlust) verursacht. Die Arrhythmien treten normalerweise unter körperlicher Belastung auf (aus irgendeinem Grund scheint Schwimmen sehr arrhythmogen zu sein) und in anderen Situationen mit hoher sympathischer Aktivität. LQT1 ist durch eine breitbasige T-Welle gekennzeichnet (Abbildung 3). LQT1 ist die häufigste Art von angeborenem LQTS.
Der Typ 2 des Long-QT-Syndroms (LQT2) wird durch eine Mutation im Kaliumkanal KCNH2 (Funktionsverlust) verursacht. Die Arrhythmien treten typischerweise bei plötzlichen Überraschungen (plötzliche Geräusche, Angst oder andere Situationen mit abruptem und plötzlichem Stress), Stress, körperlicher Aktivität oder im Schlaf auf. Die T-Welle hat eine niedrige Amplitude mit einem zusätzlichen Höcker oder einer Kerbe (Abbildung 3). Frauen mit LQT2, die sich in der postpartalen Phase befinden, haben ein sehr hohes Risiko, eine Torsade-de-Pointes-Tachykardie zu entwickeln.
Typ 3 des Long-QT-Syndroms (LQT3): wird durch eine Mutation im Natriumkanal SCN5A verursacht (führt zu erhöhten Natriumströmen). Das Risiko von Arrhythmien ist im Schlaf am höchsten. Bradykardie ist bei diesen Patienten auch stark arrhytmogen. Die ST-Strecke ist lang, die T-Welle erscheint spät und steil (Abbildung 3).
Typ 4 des Long-QT-Syndroms (LQT4): ist selten und repräsentiert 1 % aller Fälle. Die Mutation findet im ANKB-Gen statt, das ein Protein kodiert, das Membranproteine am Zytoskelett verankert. LQT3 kann mehrere Arrhythmien verursachen, wie z.B. die familiäre ventrikuläre Katecholamin-Tachykardie, Vorhofflimmern, Leitungsdefekte, Sinusknotendysfunktion und Bradykardie.
Die anderen Varianten von LQTS sind extrem selten und gehören normalerweise zu schwereren Syndromen, die mehrere Organsysteme betreffen. Diese Typen werden hier nicht diskutiert.
Schwartz-Kriterien für die Diagnose von kongenitale LQTS
Die Schwartz-Kriterien werden verwendet, um das kongenitale LQTS zu diagnostizieren. Diese Kriterien sind in Tabelle 1 aufgeführt.
Tabelle 1. Diagnosekriterien des Long-QT-Syndroms (Schwartz et al):
| EKG-Veränderungen | KRITERIEN | PUNKTE |
|---|---|---|
| QTc-Zeit | ≥480 ms | 3 |
| 460–479 ms | 2 | |
| 450–459 ms (Männer) | 1 | |
| QTc-Zeit während der 4. Minute der Erholung vom Belastungstest ≥480 ms | 1 | |
| Torsade-de-Pointes | 2 | |
| alternierende T-Welle | 1 | |
| Niedrige Herzfrequenz in Relation zum Alter (Ruheherzfrequenz unter dem 2. Perzentil) | 0.5 | |
| Klinische Geschichte | ||
| Synkope unter Stress | 2 | |
| Synkope ohne Stress | 1 | |
| Angeborene Taubheit | 0.5 | |
| Familienanamnese | ||
| Familienmitglieder mit gesichertem LQTS | 1 | |
| Unerklärlicher plötzlicher Herztod unter 30 Jahren bei unmittelbaren Familienmitgliedern | 0.5 |
Risikobewertung
- ≤1Punkt: geringe Wahrscheinlichkeit eines LQTS.
- 1.5 – 3 Punkte: intermediäre Wahrscheinlichkeit eines LQTS.
- ≥3,5 Punkte: hohe Wahrscheinlichkeit eines LQTS.
Anmerkungen
- EKG-Befunde gelten nur in Abwesenheit von Medikamenten oder Störungen, von denen bekannt ist, dass sie diese elektrokardiographischen Merkmale beeinflussen.
- QTc wird nach der Bazett-Formel berechnet, wobei QTc = QT/√RR ist.
- Es kann nur eine Synkope und Torsade-de-Pointes-Episode zählen.
- Dasselbe Familienmitglied kann unter Familienanamnese nicht in beide Kategorien fallen.
Medikamenteninduziertes Long-QT-Syndrom
Das durch Medikamente verursachte Long-QT-Syndrom ist viel häufiger als angeborene Varianten. Zu den Medikamenten, die ein Long-QT-Syndrom induzieren oder verschlimmern können, gehören Adrenalin, bestimmte Antihistaminika, Erythromyzin, Trimetoprim, Sulfonamide, Chinidin, Procainid, Disopyramid, Sotalol, Probucol, Bepridil, Difetilid, Ibutilid, Cisaprid, Ketokonazol, Itrakonazol, trizyklische Antidepressiva, Phenotiazine , Haloperidol, Indapimid, bestimmte antivirale Medikamente usw. (Tabelle 2). Die Liste der Medikamente, die LQTS verursachen, ist sehr lang und wird ständig aktualisiert. Eine vollständige Liste wird von CredibleMeds bereitgestellt (www.crediblemeds.com).
Tabelle 2. Medikamente, die ein Long-QT-Syndrom verursachen oder verschlimmern.
| KLASSE | MEDIKAMENT | ASSOZIATION | RISIKO VON TORSADE DE POINTES | WIRKUNG | KOMMENTARE |
|---|---|---|---|---|---|
| Anästhetika | Enfluran | wahrscheinlich | Medikamenten-wechselwirkungen führen zu QT-Verlängerung | ||
| Halothan | wahrscheinlich | Unspezifische Arrythmien werden in der Fachinformation genannt | |||
| Isofluran | wahrscheinlich | ||||
| Antiarrhythmika | Amiodaron | sicher | hoch | QT interval prolongation, Torsade de pointes | i.v. affects QTc less than oral; proarrhythmia infrequent. |
| Adenosin | vermutet | ||||
| Disopyramid | sicher | QT-Zeit-Verlängerung, Torsade-de-Pointes | Häufigkeit scheint niedriger als bei Chinidin zu sein | ||
| Dofetilid | sicher | hoch | QT-Zeit-Verlängerung, Torsade-de-Pointes | Proarrhythmische Wirkung 0.8 %. | |
| Flecainid | sicher | hoch | QT-Zeit-Verlängerung, Torsade-de-Pointes | Proarrhythmische Wirkung “selten”. | |
| Ibutilid | sicher | hoch | QT-Zeit-Verlängerung, Torsade-de-Pointes | Proarrhythmische Wirkung 1.7 %. | |
| Procainamid | sicher | hoch | QT-Zeit-Verlängerung, Torsade-de-Pointes | Häufigkeit scheint niedriger als bei Chinidin | |
| Propafenon | sicher | moderat | QT-Zeit-Verlängerung, Torsade-de-Pointes | Proarrhythmische Wirkung “selten”. | |
| Chinidine | sicher | hoch | QT-Zeit-Verlängerung, Torsade-de-Pointes | “Chinidin-Synkope” bei 2–6 % der Patienten. | |
| Sotalol | sicher | hoch | QT-Zeit-Verlängerung, Torsade-de-Pointes | Proarrhythmische Wirkung ~2 %. | |
| Antikonvulsiva | Felbamat | vermutet | QT-Zeit-Verlängerung, Torsade-de-Pointes | ||
| Fosphenytoin | vermutet | QT-Zeit-Verlängerung laut Hersteller. | |||
| Antidepressiva | Amitriptylin | sicher | moderat | Unspezifische EKG-Veränderungen laut Fachinformation. | |
| Citalopram | wahrscheinlich | ||||
| Desipramin | sicher | QT-Zeit-Verlängerung | Kammerflimmern, plötzlicher Herztod laut Hersteller. | ||
| Doxepin | sicher | moderat | |||
| Fluoxetin | wahrscheinlich | QT-Zeit-Verlängerung, Torsade-de-Pointes | Ventrikuläre Arrythmien bei 1 von 10 000 laut Hersteller. | ||
| Imipramin | sicher | moderat | Unspezifische Arrythmien laut Fachinformation. | ||
| Maprotilin | sicher | EKG-Veränderung; QRS laut Fachinformation. | |||
| Nortriptylin | sicher | Unspezifische Arrythmien laut Fachinformation. | |||
| Paroxetin | wahrscheinlich | Torsade-de-Pointes | Niedrigeres Risiko als bei TZA. | ||
| Sertralin | wahrscheinlich | QT-Zeit-Verlängerung, Torsade-de-Pointes | Niedrigeres Risiko als bei TZA. | ||
| Venlafaxin | vermutet | QT-Zeit Verlängerung | Arrythmierisiko von 1:1000 laut Fachinformation. | ||
| Antihistaminika | Astemizol | sicher | moderat | nicht anwendbar | |
| Clemastin | vermutet | ||||
| Diphenhydramin | vermutet | ||||
| Loratadin | vermutet | unbekannt | Verlängerung erscheint unwahrscheinlich | ||
| Terfenadin | sicher | moderat | |||
| Antiinfektiva | Clarithromycin | wahrscheinlich | moderat | QT-Zeit-Verlängerung, Torsade-de-Pointes | |
| Erythromycin | sicher | mittel bis hoch | QT-Zeit-Verlängerung, Torsade-de-Pointes | Bekannte Medikamenten-wechselwirkungen mit anderen Substanzen (z.B., Terfenadin). | |
| Fluconazol | wahrscheinlich | Risiko könnte bei i.v.-Gabe höher sein. | |||
| Foscarnet | vermutet | QT-Zeit-Verlängerung | |||
| Ganciclovir | vermutet | ||||
| Gatifloxacin | wahrscheinlich | QT-Zeit-Verlängerung | |||
| Grepafloxacin | sicher | ||||
| Halofantrin | sicher | moderat | |||
| Ketoconazol | wahrscheinlich | QT-Zeit-Verlängerung, Torsade-de-Pointes | Bekannte Medikamenten-wechselwirkungen mit anderen Substanzen (z.B., Cisaprid). | ||
| Levofloxacin | vermutet | Torsade-de-Pointes | Niedrigeres Risiko als bei vergleichbaren Substanzen. | ||
| Mefloquin | vermutet | QT mit Halofantrin. | |||
| Moxifloxacin | wahrscheinlich | QT-Zeit-Verlängerung | Niedrigeres Risiko als bei vergleichbaren Substanzen. | ||
| Pentamidin | sicher | moderat | QT-Zeit-Verlängerung, Torsade-de-Pointes | ||
| Chinin | wahrscheinlich | moderat | |||
| Sparfloxacin | sicher | ||||
| Trimethoprim-Sulfamethoxazol | vermutet | niedrig | |||
| Antipsychotika | Chlorpromazin | wahrscheinlich | Unspezifische EKG-Veränderungen und plötzlicher Herztod laut Hersteller. | ||
| Clozapin | vermutet | ||||
| Haloperidol | sicher | moderat | QT-Zeit-Verlängerung, Torsade-de-Pointes | ||
| Mesoridazin | sicher | QT-Zeit-Verlängerung, Torsade-de-Pointes | |||
| Pimozide | sicher | QT-Zeit-Verlängerung, Torsade-de-Pointes | Medikamenten-wechselwirkungen führen auch zu QT-Verlängerung. | ||
| Quetiapin | vermutet | QT-Zeit-Verlängerung | |||
| Risperidon | vermutet | QT-Zeit-Verlängerung | Plötzlicher Herztod laut Hersteller. | ||
| Sertindole | vermutet | moderat | |||
| Thioridazin | sicher | moderat | QT-Zeit-Verlängerung, Torsade-de-Pointes | ||
| Ziprasidon | sicher | QT-Zeit-Verlängerung | |||
| Onkologische Medikamente | Arsentrioxid | sicher | QT-Zeit-Verlängerung, Torsade-de-Pointes | ||
| Tamoxifen | vermutet | QT-Zeit-Verlängerung | Überdosis-Situationen. | ||
| Kardiovaskuläre Medikamente | Bepridil | sicher | moderat | QT-Zeit-Verlängerung, Torsade-de-Pointes | QTc um ~8 % verlängert. |
| Indapamid | vermutet | QT-Zeit-Verlängerung | |||
| Isradipin | vermutet | QT-Zeit-Verlängerung laut Hersteller. | QTc um ~3 % verlängert. | ||
| Mibefradil | vermutet | ||||
| Moexipril-Hydrochlorothiazid | vermutet | QT-Zeit-Verlängerung laut Hersteller. | |||
| Nicardipin | vermutet | QT-Zeit-Verlängerung laut Hersteller. | |||
| Probucol | sicher | ||||
| Gatrointestinale Medikamente | Cisaprid | sicher | moderat | QT-Zeit-Verlängerung, Torsade-de-Pointes | |
| Octreotid | vermutet | QT-Zeit-Verlängerung | |||
| Droperidol | sicher | sicher | QT-Zeit-Verlängerung, Torsade-de-Pointes | ||
| Dolasetron | vermutet | QT-Zeit-Verlängerung laut Hersteller. | |||
| Migränemedikamente | Naratriptan | wahrscheinlich | QT-Zeit-Verlängerung | ||
| Sumatriptan | wahrscheinlich | QT-Zeit-Verlängerung | |||
| Rizatriptan | wahrscheinlich | Arrhythmierisiko von 1:1000. | |||
| Zolmitriptan | wahrscheinlich | QT-Zeit-Verlängerung | |||
| Weitere Substanzen | Amantadin | gering | |||
| Epinephrin | vermutet | ||||
| Levomethadyl | wahrscheinlich | QT-Zeit-Verlängerung, Torsade-de-Pointes | |||
| Methadon | wahrscheinlich | Synkope laut Hersteller. | |||
| Salmeterol | vermutet | QT-Zeit-Verlängerung laut Hersteller. | |||
| Tacrolimus | vermutet | ||||
| Tizanidin | wahrscheinlich | QT-Zeit-Verlängerung | Arrhythmierisiko von 1:1000. |
Management des Long-QT-Syndroms (LQTS)
Hämodynamisch instabile Torsade-de-Pointes-Tachykardie
Eine Torsade-de-Pointes-Tachykardie, die zu einer Synkope führt, wird mit Defibrillation behandelt. Beginnen Sie mit 150 J (biphasischer Schock) und erhöhen Sie bei jedem folgenden Schock um 50 J. Kammerflimmern und Herzstillstand werden nach dem Reanimations-Algorithmus behandelt.
Hämodynamisch stabile Torsade-de-Pointes-Tachykardie
Die Behandlung von Torsade-de-Pointes-Tachykardien ist bei angeborenen und erworbenen LQTS ähnlich. Torsade-de-Pointes-Tachykardien sind paroxysmal, was bedeutet, dass die Arrhythmie intermittierend auftritt und selbstlimitierend ist. Sie neigt zu Rezidiven, auch nach erfolgreicher Defibrillation. Es besteht immer das Risiko von Kammerflimmern, weshalb ein Defibrillator in der Nähe sein muss und Reanimationsbereitschaft notwendig ist.
Behandlungsalgorithmus
- Alle Arzneimittel/Medikamente, die die Arrhythmie verursachen oder verschlimmern können, müssen sofort gestoppt werden.
- Magnesiuminfusion (unabhängig vom Magnesiumspiegel im Blut): 1 Gramm Magnesium über 60 Sekunden intravenös verabreicht. Dies kann nach 5 bis 10 Minuten wiederholt werden. Wenn eine kontinuierliche Infusion erforderlich ist, beträgt die Dosis 5—10 mg/min.
- Kaliuminfusion: nur notwendig, wenn der Patient eine Hypokaliämie hat.
- Bradykardie muss korrigiert werden: Bradykardie kann eine Torsade-de-Pointes-Tachykardie auslösen und verschlimmern. Um Bradykardie zu korrigieren, stehen folgende Optionen zur Verfügung:
- Atropin i.v. 1–2 ml 0.5 mg/ml.
- Isoprenalin (Isoproterenol) 0.01 μg/kg/min, dies wird titriert, bis die Bradykardie endet. Beachten Sie, dass Isoprenalin vorsichtig verabreicht werden muss, da es betaadrenerge Rezeptoren aktiviert und so die Arrhythmie verschlimmern kann. Bei kongenitalem LQTS ist Isoprenalin kontraindiziert, da das Risiko für Kammerflimmern hoch ist. Daher darf Isoprenalin nur bei erworbenem LQTS und vorübergehend verwendet werden, bis ein Herzschrittmacher verfügbar ist.
- temporärer transkutaner/transvenöser Schrittmacher. Die Schrittmacherelektrode sollte in den Vorhöfe platziert werden und die Frequenz sollte auf 90 Schläge pro Minute eingestellt werden. Die Frequenz kann schrittweise erhöht werden, bis die Arrhythmie sistiert.
Die Begründung für Atropin-, Isoprenalin- und Schrittmacher-Therapie ist ganz einfach: Diese drei Interventionen erhöhen alle die Herzfrequenz, was das QTc-Intervall verringert und somit die Torsade-de-Pointes beendet.
Langzeitbehandlung des erworbenen Long-QT-Syndroms
Nach Absetzen der das Syndrom verursachenden Medikamente ist keine Behandlung erforderlich.
Langzeitbehandlung des kongenitalen Long-QT-Syndroms
Betablocker sind bei angeborenem LQTS sehr effektiv. Die Sterblichkeit wird drastisch reduziert, wenn das richtige Medikament und die richtige Dosis verabreicht werden. Propranolol (normalerweise ausreichend mit 3 mg/kg/Tag) und Nadolol (normalerweise ausreichend mit 1 mg/kg/Tag) sind die wirksamsten Medikamente. Metoprolol hat eine nachgewiesene Wirkung, ist aber weniger wirksam als Propranolol und Nadolol. Es gibt keine Studien zu Atenolol, das daher nicht empfohlen werden kann. Patienten mit ausgeprägter Bradykardie sollten keine Betablocker erhalten, da das Risiko besteht, Torsade-de-Pointes zu provozieren.
Ein künstlicher Herzschrittmacher kann erforderlich sein, wenn Betablocker in Maximaldosis nicht ausreichen. Wenn eine Schrittmacherbehandlung ebenfalls nicht ausreicht, kann eine Sympathektomie in Betracht gezogen werden. Sympathektomie bedeutet, dass sympathische Nervenganglien (des Thorax) operativ entfernt werden, was zur Beseitigung der adrenergen Stimulation des Herzens führt. Dies ist eine effektive Methode, erfordert jedoch eine Operation.
ICD-Therapie des Long-QT-Syndroms (LQTS)
Ein ICD (implantierbarer Kardioverter/Defibrillater, engl. Intracardial Cardiac Defibrillator) wird in den folgenden Fällen verwendet:
- Bei Patienten, die einen Herzstillstand erlitten haben.
- Bei Patienten, die trotz optimaler Behandlung eine Synkope erlebt haben (Betablocker in Maximaldosis, Herzschrittmachertherapie und möglicherweise Sympathektomie).
- Wenn die Anamnese sehr besorgniserregend ist und das QTc-Intervall >550 ms beträgt. Das Auftreten von alternierenden T-Wellen und Sinuspausen bekräftigt dies.
Short-QT-Syndrom (Short QT syndrome, SQTS)
Das Short-QT-Syndrom ist extrem selten, kann jedoch eine polymorphe ventrikuläre Tachykardie verursachen. Es ist definiert als QTc-Zeit <0,35 s. Beachten Sie, dass Hyperkalzämie und Digitalisglykoside auch zu einer QTc-Zeit-Verkürzung führen können.
